Přírodovědecká fakulta Masarykovy univerzity
prof. RNDr. Jiřina Hofmanová, CSc.
Mutace, genetická nestabilita
Význam mutací u nádorových onemocnění
Mutace je definována jako kvalitativní nebo kvantitativní změna v genetické informaci obsažené v DNA. Je považována za nevratnou (ireverzibilní) změnu a může být indukována fyzikálními, chemickými či jinými faktory.
Mutace mohou vznikat na úrovni genů nebo chromozómů. Jeden specifický gen může být změněn přidáním, ztrátou nebo záměnou jedné baze. Protein kódovaný tímto genem pak může být změněn s ohledem na strukturu a funkci (obr. 2-1).
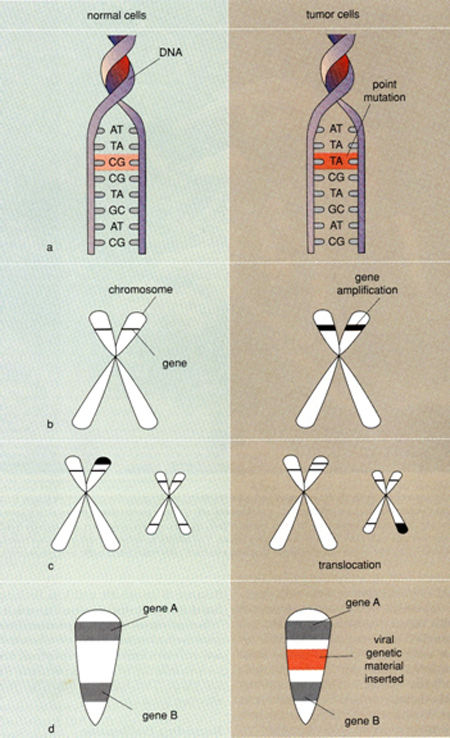
Obr. 2-1 Genetické změny a disorganizace chromozómů v nádorových buňkách
(Van Noorden C.J.F. et al., American Scientist 1998 : 130)
Jestliže mutace zahrnuje změnu v počtu chromozómů v buňce (nondisjunkce, polyploidizace), tak potom, i když jednotlivé geny jsou normální, jejich počet může narušit jejich funkci. Změny v uspořádání chromozómů způsobené delecí nebo translokací části jiných chromozómů také mohou způsobit jak mutace genů, tak abnormální expresi genů umístěných na takovémto chromozómu. Není pochyb, že mutace hrají u nádorových onemocnění důležitou úlohu. Jak onkogeny a nádorově supresorové geny tak i abnormální chromozómy a počty chromozómů jsou spojeny s nádorovými buňkami (obr. 2-2).
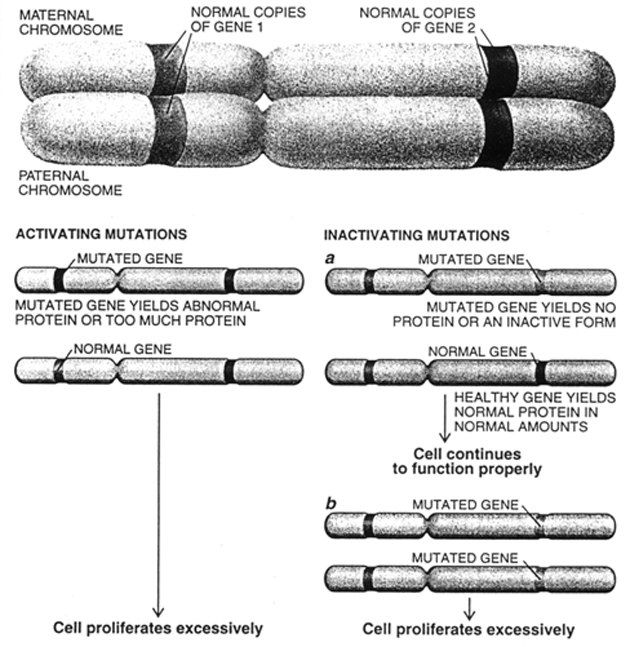
Obr. 2-2 Geny se dědí v odpovídajících párech
(Cavence E.K. and White R.L., Scientific American 1995 : 50)
Jako mutageny mohou působit různé chemické látky i fyzikální faktory (např. ionizující či UV záření), ale i viry, které indukují v buňkách mutace. Kromě toho existují dědičné mutace (Xeroderma pigmentosum, Wilmsův nádor, albinismus, Lynchův syndrom atd.) předurčující jedince k určitým typům nádorů.
Důležité je, uvědomit si, že mutagenita se nerovná karcinogenitě. Ze studií vyplývá, že ačkoliv mutageny mají vysokou pravděpodobnost být i karcinogeny (84 %), může docházet k falešným predikcím, protože řada nekarcinogenních látek jsou rovněž mutageny. Naopak také mnoho nemutagenních chemikálií může být karcinogenní.
Chemikálie jsou označovány jako karcinogeny, jestliže v exponované lidské populaci je vyšší frekvence nádorů než v neexponované populaci nebo jestliže se objevují nádory u zvířat, kterým byla chemikálie podávána.
Existuje také řada látek či faktorů, které fungují jako kokarcinogeny, tím, že např. zvyšují hladiny buněčných enzymů, které aktivují karcinogeny.
Antikarcinogeny se zase chemicky váží na karcinogen, odbourávají jej, tlumí enzymy aktivující karcinogeny nebo obsadí cílové místo (kompetitivní inhibice).
Mutace jsou výsledkem různých typů mechanizmů. Některé bodové vznikají následkem chyb v reparaci poškozené DNA při replikaci DNA s nereparovanými poškozeními. Na druhé straně některé mutace mohou vzniknout při replikaci normální DNA s defektivní DNA polymerázou nebo normální DNA polymerázou, která je inhibovaná.
Změny v počtu chromozómů jsou pravděpodobně způsobeny faktory, které neovlivňují přímo DNA, ale ovlivňují molekuly zodpovědné za segregaci chromozómů během buněčného dělení. Chromozomální přestavby jsou pravděpodobně způsobeny jak faktory poškozujícími DNA, tak i enzymy nutné pro její stabilitu.
Mutace jsou nejen znakem nádorů, ale jsou také zásadní pro jejich vývoj. Normální frekvence mutací 10-7–10-8 / nukleotid / buněčné dělení. Zvýšená frekvence mutací podporuje karcinogenezi. Molekulární techniky dnes umožňují zkoumat lidský genom od chromozómů až po sekvence nukleotidů a odhalovat tak stále více a více mutací v nádorových buňkách.
Genom nádorových buněk je nestabilní a tato nestabilita vyúsťuje v kaskádu mutací, z nichž některé umožňují nádorovým buňkám obejít regulační procesy, které kontrolují lokalizaci buňky, její dělení, adaptaci a smrt. Genetická nestabilita je manifestována velkou heterogenitou buněk v každém nádoru, přispívá k jejich progresi. I k rozvoji rezistence k chemoterapeutikům.
Poškozování a reparace DNA
Každý nádorový fenotyp obsahuje specifické mutace v kritických genech. Tyto mutace vznikají buď kopírováním nereparovaných poškození v DNA, nebo chybami během syntézy DNA (obr. 2-3).
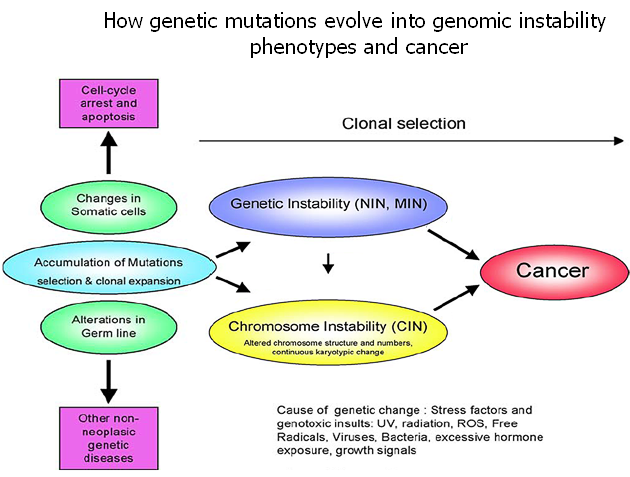
Obr. 2-3 Spojení genotoxického stresu a genomové nestability
(Jefford Ch.E., Irminger-Finger I., Crit Rev Oncol/Hematol 2006 : 1)
Současné studie předpokládají dva hlavní mechanizmy vzniku mutací v nádorových buňkách:
- deficit v reparaci DNA (kopírování nereparovaných poškození v DNA nebo chyby během syntézy DNA)
- deficit v rozdělování chromozómů při buněčném dělení
Zdrojem nepřesností při replikaci DNA jsou chyby vzniklé při DNA polymeraci (tj. kvalita DNA polymeráz a souvisejících „proofreadingových“ procesů) a chyby v systémech reparace DNA. Chyby purifikovaných DNA polymeráz in vitro se pohybují od 5000 pro polymerázu beta, která syntetizuje krátké úseky, po 10 mil. pro polymerázu delta a eta účastnící se replikace DNA. U nádorů nebyly prokázány defekty v DNA polymerázách, ale byly prokázány defekty ve dvou hlavních systémech reparace DNA.
Nukleotidová excizní oprava
(„nucleotide-excision repair“ – NER) – s ní spojená nestabilita („NER-associated instability“ – NIN)Oprava špatného párování
(„mismatch repair“ – MMR) – s ní spojená mikrosatelitová nestabilita (MIN) (obr. 2-4)
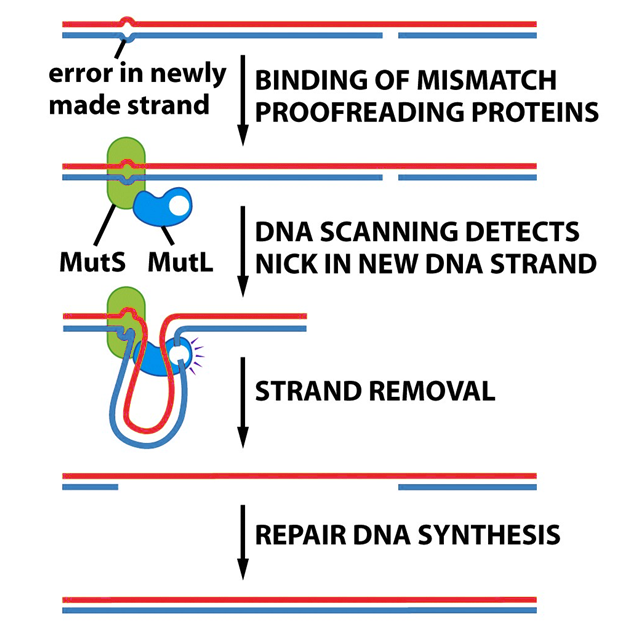
Obr. 2-4 Oprava špatného párovaní bazí
(Figure 12.8c. Weinberg R. A., The Biology of Cancer (© Garland Science 2007))
V buněčné DNA dochází ke stálému poškozování a resyntéze. DNA je poškozována jak environmentálními tak endogenními zdroji. Mnoho z těchto látek jsou mutageny a řada z nich i karcinogeny. Poškození DNA chemikáliemi spadá do dvou kategorií:
- ty, které produkují velké rozsáhlé adukty a jsou reparovány excisí nukleotidů a
- ty, které způsobují malé změny reparované excisní reparací bazí, jako např. alkylační činidla, která přidávají metyl- a etylskupiny do nukleotidů.
Bylo prokázáno, že kromě chemikálií existuje řada přírodních látek v potravě, které také mohou poškozovat DNA. Konzumace těchto látek je pravděpodobně daleko vyšší než expozice průmyslově produkovanými chemikáliemi.
Buněčné metabolické procesy rovněž produkují reaktivní chemické produkty se schopností poškozovat DNA a tak produkovat spontánní mutace. Dokonce i voda má schopnost poškozovat DNA, když způsobí hydrolýzu glykosilických můstků a tvorbu mutagenních abazických míst. Odhaduje se, že každá buňka je každý den podrobena 10 000 depurinací. Poškození DNA reaktivními kyslíkovými radikály (ROS) se vyskytuje přibližně se stejnou frekvencí a poškozené baze např. 8-oxo-deoxyguanosine narušují kód. Ačkoliv je deaminace cytosinu na tymidin méně častá, způsobuje změnu sekvence nukleotidů. Vzhledem k vysoké frekvenci poškození je pravděpodobné, že významná část poškození unikne reparaci a produkuje mutace během replikace této nereparované DNA DNA polymerázami. Týká se to zejména drobných změn. Rozsáhlé adukty je obtížné obejít a indukce mutací závisí na indukci dalších mechanizmů zahrnujících chybnou syntézu DNA.
Nukleotidové sekvence v buněčné DNA jsou udržovány v homeostatické rovnováze, kdy růst poškození DNA nebo snížení reparace vedou ke zvýšené frekvenci mutací (obr. 2-5).
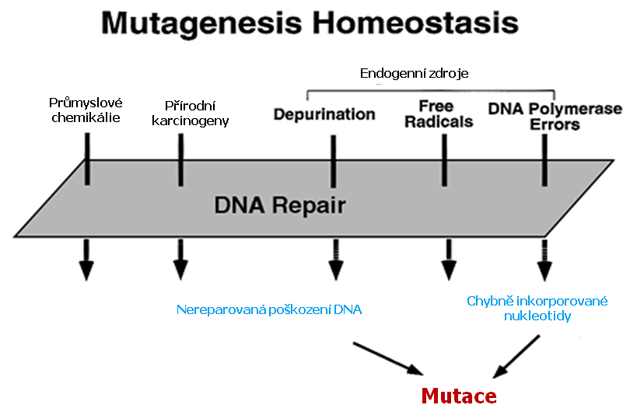
Obr. 2-5 Faktory vedoucí k akumulaci mutací v nádorových buňkách
(Loeb K. R. and Loeb L. A., Carcinogenesis 2000 : 379)
Obrovské množství chromozomálních aberací a heterogenita nádorů předpokládají, že nádorové buňky jsou geneticky nestabilní. K této nestabilitě přispívají v zásadě a nezávisle dva překrývající se mechanizmy:
- vznik mutovaného fenotypu nádorových buněk založený na vzrůstajícím počtu chyb v syntéze DNA během replikace. Tyto chyby vznikají v důsledku mutací v DNA polymerázách, takže vnášejí chyby nebo mutací v DNA reparačních proteinech, které jsou tak defektní. Některé z těchto mutací nastávají v genech důležitých pro udržování genetické stability
- akumulace mutací založená na postupných vlnách klonální selekce. Tento model vytvořil na základě analýzy sekvence chromozomálních aberací v nádorech Nowell v r. 1976 (obr. 2-6).
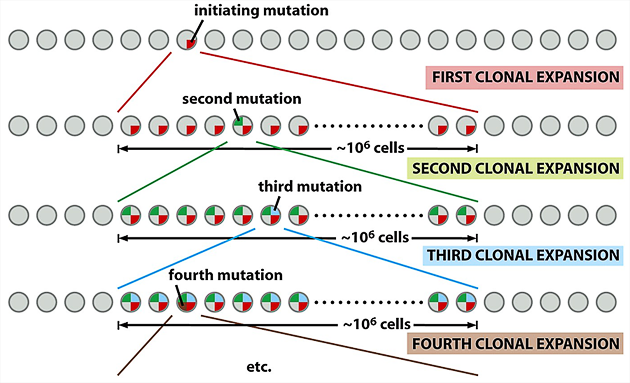
Obr. 2-6 Hromadění, selekce a klonální expanze mutací
(Figure 11.12. Weinberg R.A., The Biology of Cancer (© Garland Science 2007))
Mikrosatelity
Bylo zjištěno, že velké množství mutací (např. až sto tisíc u nádoru kolonu) se nachází v tzv. mikrosatelitech – repetitivních sekvencích mezi geny. Nyní se ukazuje, že repetitivní sekvence jsou i uvnitř genů a jsou u nádorových buněk zkracovány nebo prodlužovány s vysokou frekvencí. Tak dochází patrně k inaktivaci nádorově supresorových genů během rozvoje nádorů. Vysoká hladina nestability mikrosatelitů se vyskytuje zejména u tzv. HNPCC – dědičný nepolypózní nádor kolonu. Dědí se zde mutace v jedné alele genů zahrnutých v opravě nesprávných spojení v DNA ("mismatch repair") a dochází s vysokou frekvencí ke ztrátě druhé divoké alely. Lokus zodpovědný za HNPCC byl zmapován do oblasti 2p16 a 3p21. V roce 1993 bylo poprvé ukázáno, že HNPCC může souviset s defekty MMR a bylo potvrzeno, že „odpovědnými“ geny jsou MSH2 na chromozómu 2 a MSH1 na chromozómu 3. MSH2 a MSH1 odpovídají za 90 % vrozených mutací HNPCC, další jsou PMS1, PMS2 a MSH6.
U jiných nádorů, u kterých dochází k expanzi nebo deleci repetitivních sekvencí, je snížená reparace DNA způsobena metylací společně s redukovanou expresí některého z genů pro "mismatch" reparaci. Na nestabilitě mikrosatelitů se ve velké míře podílejí ROS, takže se uvažuje o terapeutickém využití látek působících jako antioxidanta.
Nádory vykazující nestabilitu mikrosatelitů obsahují často změny v délce repetitivních sekvencí uvnitř řady genů spojených s nádory jako je APC, IGF, TGF-beta, hMSH3, hMSH6.
Chromozomální nestabilita
Genetická nestabilita nádorových buněk se projevuje nejen na úrovni nukleotidů vznikajícími bodovými mutacemi, ale také na úrovni celých chromozómů translokacemi, delecemi, amplifikacemi a aneuploidií. Oba typy nestability vedou k mutantnímu fenotypu prostřednictvím změněné exprese proteinů, funkcí nebo efektem genové dávky.
Amplifikace (zmnožení) genů. Téměř všechny nádory prsu a vaječníků studované s využitím srovnávací genomové hybridizace obsahují řadu změn v počtu genových kopií. Genová amplifikace se objevuje u některých typů nádorů vyšších stádií a může souviset s rezistencí k chemoterapeutikům (N-myc, erb a ras). Amplifikace se objevují v pozdních stádiích maligní transformace, jsou spojeny s agresivně rostoucími nádory a signalizují nepříznivý prognostický vývoj.
Aneuploidie, změna v počtu chromozómů, je vlastností řady nádorů. Protože každý chromozóm obsahuje tisíce genů, je udivující, že změny v počtu chromozómů jsou slučitelné s viabilitou buněk. Specifické chromozomální výměny se vyskytují ve vysoké frekvenci a jsou detekovány v určitých nádorech. Obecně existuje pozitivní korelace mezi počtem chromozomálních změn v nádoru a maligním potenciálem tohoto nádorového onemocnění.
Je však poměrně málo známo o tom, jak se aneuploidie vyvíjí nebo o selektivních výhodách jaké přináší nádorům. Aneuploidie může vzniknout fragmentací chromozómů, translokací, amplifikací nebo nondisjunkcí. Progresivní růst aneuploidie je raným počátečním dějem vedoucím ke genetické nestabilitě a je nezávislý na akumulaci jiných typů mutací (obr. 2-7).
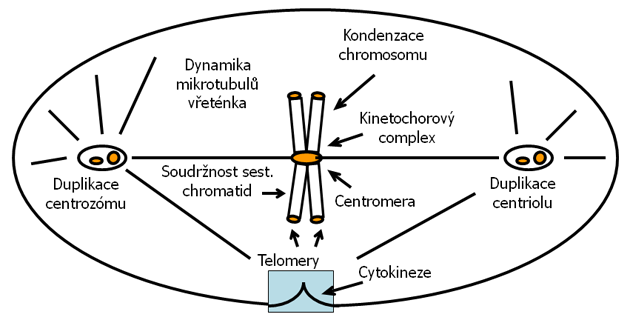
Obr. 2-7 Potenciální mitotické cíle vedoucí k aneuploidii
(podle Sen S., Curr Opin Oncology 2000 : 82)
Předpokládá se, že nejdříve vznikají tetraploidní mezistupně po defektní mitóze nebo endoreduplikaci. Po přechodné zástavě mitózy v přítomnosti poškození vřeténka, jsou některé nádorové buňky schopny obnovit buněčný cyklus vstupem do dalšího kola syntézy DNA, což vede k endoreduplikaci. Počínají si tak zejména buňky, které postrádají funkční produkty genů kontrolujících přechod fáze G1/S jako jsou p53, pRb, p16, p21 i buňky se zvýšenou expresí onkogenu myc. Úloha defektních kontrolních mitotických bodů při vzniku aneuploidie však není ještě zcela jasná
Předpokládá se, že existují dva základní mechanizmy vzniku genetické nestability (obr. 2-8):
- Mechanizmus zahrnující mutace v genech pro opravu nesprávných spojení DNA (mismatch repair) a manifestují se nestabilitou mikrosatelitů. Je zapotřebí mutace v obou alelách.
- Mechanizmus manifestující se fragmentací chromozómů nebo duplikací či delecí celých chromozómů. Stačí pouze jedna mutace, protože fenotyp nestability chromozómů má dominantní charakter.
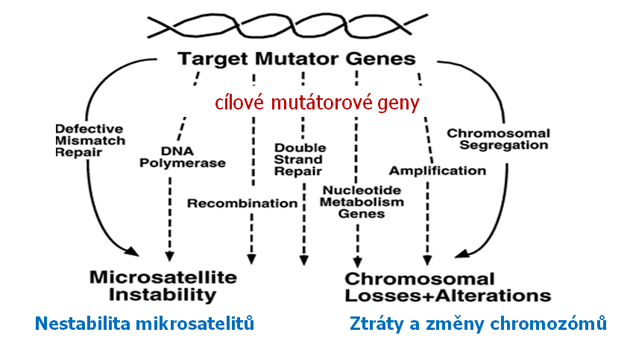
Obr. 2-8 Různé dráhy vedoucí ke vzniku mutovaného fenotypu nádorové buňky
(Loeb K. R. and Loeb L. A., Carcinogenesis 2000 : 379)
Důležitým procesem při vývoji nádorů je rovněž ztráta heterozygotnosti („lost of heterozygosity“, LOH). Buňky, které nesou jednu mutovanou alelu nádorového supresoru, ztrácejí delecí velkou část chromozómu, který nese funkční alelu. V oblastech s vysokou frekvencí LOH často leží geny nádorových supresorů. Buňky, které nesou jednu mutovanou alelu nádorového supresoru (např. retinoblastoma- RB gen), ztrácejí funkční alelu (obr. 2-9).
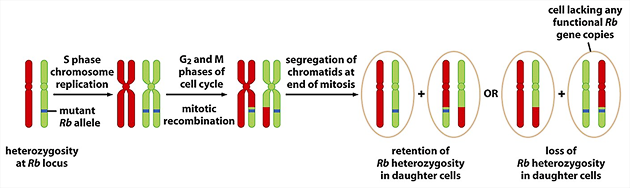
Obr. 2-9 Ztráta heterozygotnosti
(Figure 7.8. Weinberg R. A., The Biology of Cancer (© Garland Science 2007))
Předpokládá se proto, že mutovaný fenotyp je manifestován pravděpodobněji nestabilitou chromozómů než bodovými mutacemi. Souvisí to však s tím, že chromozomální změny jsou daleko snadněji detekovatelné než náhodné bodové mutace.
S růstem počtu mutací přispívá ke genetické nestabilitě jev klonální selektivity. Jak se nádor rozrůstá, střetává se s řadou překážek omezujících jeho růst. Je to vliv okolních tkání, omezená výživa a přístup kyslíku, potřeba růstových faktorů, nedostatečné zásobování krví atd. Každá z těchto překážek může být překonána v důsledku mutací, které poskytují růstovou výhodu a ustanovují novou klonální populaci. S každým kolem selekce dochází také ke vzniku dalších mutací (obr. 2-10).
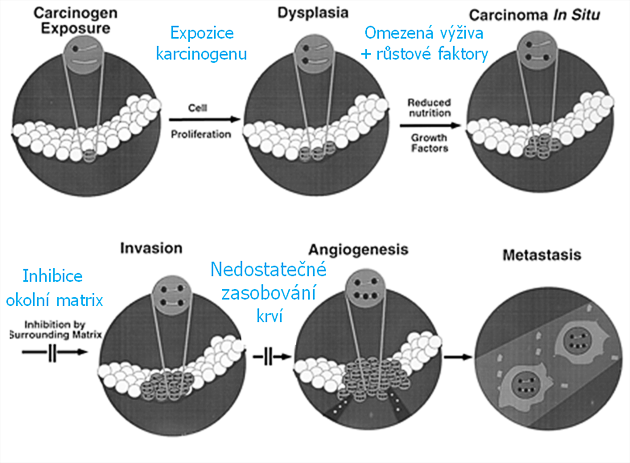
Obr. 2-10 Vývoj nádoru založený na selekci mutovaného fenotypu
(Loeb K. R. and Loeb L. A., Carcinogenesis 2000 : 379)
Díky genetické nestabilitě dochází během času k akumulaci mutací buď v gametách, nebo somatických buňkách. Somatické nebo dědičné genetické změny zahrnuté v genetické nestabilitě postihují funkce jako je reparace DNA a kontrolní body buněčného cyklu a mitózy, což předchází zvýšenou telomerázovou aktivitu. Somatické buňky však vyžadují pro rozvoj nádoru ještě dysfunkci tzv. „gatekeepers“. Mutace umožňující genomovou nestabilitu jsou v gametách i somatických buňkách selektovány.
„Gatekeepers“ jsou recesivní geny, které přímo regulují (limitují) růst nádorů buď inhibicí jejich růstu nebo indukcí jejich smrti.
„Caretakers“ jsou geny, jejichž inaktivace navozuje genetickou nestabilitu a ta pouze nepřímo indukuje růst nádorů zvyšováním mutační rychlosti.
Geny jako jsou APC, či p53 fungují zároveň jako „gatekeepers“ i „caretakers“.
Kontrolní body buněčného cyklu, apoptóza a mutovaný fenotyp
V eukaryotické buňce existují kontrolní body, které zastavují replikaci DNA a umožňují reparaci. Při mutaci genů kontrolujících např. přechod G1/S buněčného cyklu je umožněna replikace nereparované DNA a výsledkem je zvýšená mutageneze.
V průběhu buněčného cyklu existuje několik kontrolních bodů, kde je monitorována reparace poškození před vstupem do následující fáze (obr. 2-11). Při aktivaci se v těchto bodech cyklus přechodně zastavuje, aby mohla být poškození reparována. Eliminace těchto kontrolních bodů vede k vývoji mutovaného fenotypu. Např. mutace v p53 způsobuje absenci kontroly poškození DNA a umožňuje nádorové buňce replikovat tuto poškozenou DNA a akumulovat mutace v dceřinných buňkách, což vede ke genomové nestabilitě. V případě vzniku nereparovatelného poškození se normálně spouští apoptóza, která zabrání rozšíření mutací (obr. 2-12). Řada nádorů však obsahuje mutace, které zpožďují nebo zabraňují apoptóze a tak podporují přežití geneticky nestabilních maligních buněk (obr. 2-13).
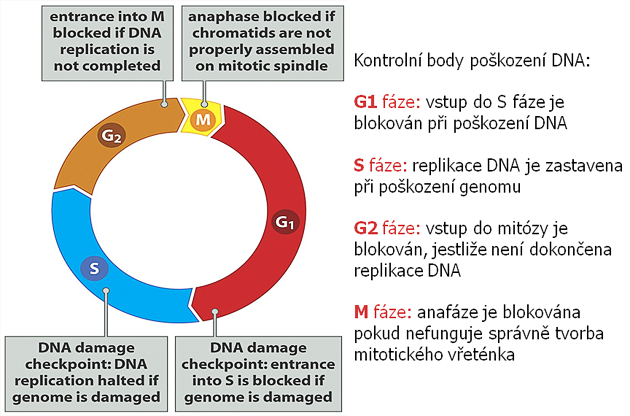
Obr. 2-11 Kontrolní body buněčného cyklu
(Figure 8.4. Weinberg R.A., The Biology of Cancer (© Garland Science 2007))
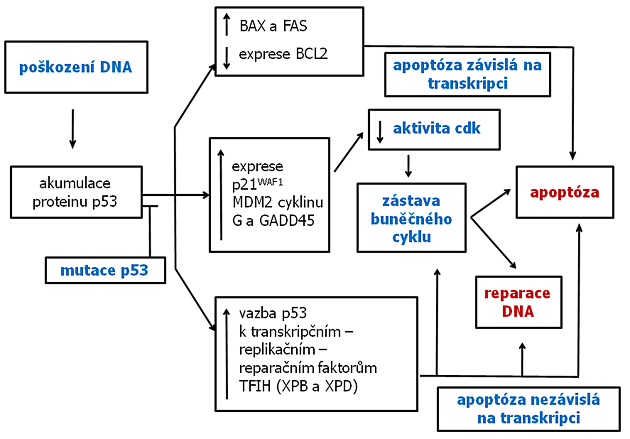
Obr. 2-12
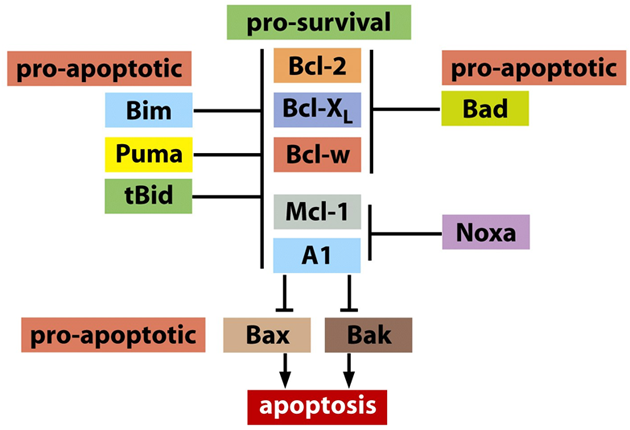
Obr. 2-13 Proteiny spouštějící nebo blokující apoptózu
(Figure 9.27c. Weinberg R.A., The Biology of Cancer (© Garland Science 2007))
Genetika vs. Enviromentální faktory
Při vzniku nádorů působí dva typy mechanizmů – environmentální faktory a genetické vybavení jedince.
V některých případech je primární environmentální faktor, který způsobí vznik nádoru u "normálního" jedince. Avšak i v tomto případě jsou zasaženy geny, protoonkogeny a nádorově supresorové geny. Kromě toho existují další geny, které mohou způsobit větší či menší citlivost (susceptibility) jedince k environmentálním faktorům.
Předpokládáme-li, že všechny nádory jsou výsledkem iniciační, promoční a progresívní fáze karcinogeneze, měly by existovat geny, které:
- buď chrání, nebo predisponují protoonkogeny nebo nádorově supresorové geny k aktivaci nebo inaktivaci,
- selektivně podporují nebo suprimují růst a expanzi iniciované buňky,
- zabraňují nebo zvyšují možnosti získání genetické/epigenetické nestability iniciované buňky, které zapříčiňují její malignitu.
Příkladem jsou některé lidské dědičné syndromy (obr. 2-14):
Albinismus
Jedinci s tímto syndromem nemají melaninovou pigmentaci, která chrání kůži nebo oči před škodlivými účinky UV záření. Mají normální reparační mechanizmy DNA, ale množství poškození je větší než je tento systém schopen zvládnout. Nereparovaná poškození DNA fungují jako substrát pro vznik mutací v protoonkogenech nebo nádorově supresorových genech. Syndrom je klasifikován jako typ náchylný ke vzniku nádorového onemocnění. Protože poškození DNA je velké, mnoho buněk umírá a smrt kožních buněk stimuluje tzv. kompenzační – regenerační růst buněk přežívajících. V tomto procesu pak vzniká možnost podpory dělení iniciovaných buněk. Klony iniciovaných buněk dále exponované UV vykazují zvýšenou pravděpodobnost dalších mutačních (genetických) změn, při jejichž hromadění dochází k postupu populace buněk do progresívního stadia.Xeroderma pigmentosum
Genetický syndrom, který také předurčuje jedince k rakovině kůže, avšak na jiném principu. Jedinci jsou nositeli genu, který neumožňuje reparovat poškození DNA indukované UV. Výsledkem jsou nereparovaná poškození DNA, která vedou ke smrti buněk nebo k mutacím. Podobně jako u albínů dochází ke kompenzační hyperplazii při náhradě odumřelé tkáně. Iniciovaná buňka kůže proliferuje, ale nediferencuje. Vytváří klon iniciovaných buněk (papilom) citlivý k indukci dalších genetických/epigenetických změn a k progresivnímu rozvoji.

Obr. 2-14 Lidské dědičné syndromy způsobené dědičnými defekty v reparaci DNA
(Figure 12.1. Weinberg R. A.,The Biology of Cancer (© Garland Science 2007))
Jedinci s albinismem nebo XP však nejsou odsouzeni k nádorovému onemocnění. Jestliže je pokožka chráněna před UV, nemusí být vyvoláno.
Při nadměrném působení UV mohou být postiženi i „normální“ jedinci. Reparační systém je přetížen a vzniká poškození DNA vedoucí opět k mutacím a ke smrti buněk podobně jako v předchozích případech. Jedinci s větší pigmentací jsou lépe chráněni před škodlivými účinky UV.
Existuje řada environmentálních faktorů a genů, které předurčují nebo chrání jedince před vznikem nádorů. Existuje řada chemikálií buď exogenních (dieta, životní styl, léčiva, polutanty) nebo endogenních (hormony, růstové faktory), které nepoškozují DNA a nejsou mutagenní. Jsou to negenotoxicky působící látky (faktory), které mohou působit jak promoční stimulací proliferace iniciované buňky, tak supresí apoptózy. Jedinci, kteří jsou normálně exponováni a akumulují iniciované buňky (což se normálně děje s přibývajícím věkem), ale kteří jsou exponováni abnormálnímu množství promočních látek v těle v důsledku genetického defektu, jsou označování jako „promotion-prone“.
Vyloučení působení nádorových promotorů může u normálních jedinců snížit vznik nádorů přesto, že dojde k iniciaci. Naopak pravidelná a chronická expozice dostatečnému množství nádorového promotoru zvyšuje riziko vzniku nádoru. Podobně jako existují antiiniciátorové geny a látky, tak existují i geny a látky působící antipromočně.
Kontrolní otázky k tématu
Jak je definována mutace?
Na jaké úrovni mohou mutace vznikat?
V jakém smyslu může být určitý protein změněn mutací?
Jaký je rozdíl mezi aktivační a inaktivační mutací?
Jaké jsou 2 hlavní mechanizmy vzniku mutací v buňkách?
Poruchy jakých typů reparace DNA jsou zásadní při vývoji nádorů?
Jakou úlohu hrají při vzniku mutací DNA polymerázy?
Jak vzniká genetická nestabilita a které mechanizmy k ní přispívají?
Co jsou to tzv. „gatekeeper“ a „caretaker“ geny?
Kde se nacházejí hlavní kontrolní body buněčného cyklu? Vyjmenuj nejdůležitější molekuly spojené s jejich regulací.
Jakou roli hraje v karcinogenezi apoptóza?
Co jsou to mikrosatelity a jakou úlohu hrají v nádorech?
Co je to genová amplifikace a její význam.
Co je aneuploidie a co k ní přispívá?
Jakou roli hraje ztráta heterozygotnosti u nádorového onemocnění?
Vysvětlete pojem klonální selektivita a jeho význam v rozvoji nádoru?
Které dva základní vlivy se uplatňují při vzniku a rozvoji nádorového onemocnění?
Charakterizujte lidské dědičné syndromy predisponující k nádorům způsobeným UV zářením.
Jaký je mechanizmus vzniku nádorů u takto postižených jedinců?
V jakých buňkách se nacházejí mutace v případě dědičných nádorů?
ÚEB Biol, Přírodovědecká fakulta, Masarykova univerzita |
Návrat na úvodní stránku webu, přístupnost |
| Servisní středisko pro e-learning na MU
| Fakulta informatiky Masarykovy univerzity, 2012–2013
Centrum interaktivních a multimediálních studijních opor pro inovaci výuky a efektivní učení | CZ.1.07/2.2.00/28.0041
